
在线客服

在线客服
医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。不同种类的医疗器械在人体中使用时风险程度具有差异,为了便于监管,我国对医疗器械按照风险程度实行分类,具体分类见下表。
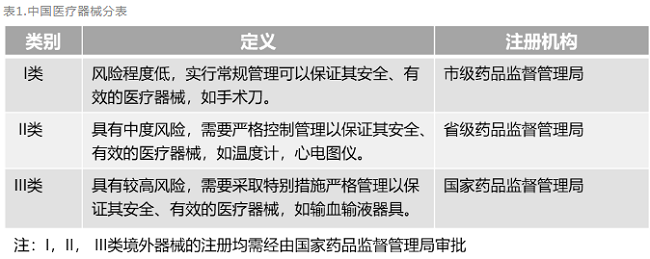
目前,在中国涉及医疗器械注册的主要是由国家药品监督管理局(NMPA)下的医疗器械技术审评中心(CMDE)负责。CMDE下设质量管理部,合规部,项目管理部,审评部,临床与生物统计部等部门,负责起草、拟订和修订医疗器械的行业标准、生产质量管理规范,制定医疗器械产品分类管理目录,开展注册审批和监督。中国目前医疗器械上市准入的法规依据是《医疗器械注册管理办法》。根据该法规,针对第Ⅰ类器械,境内生产企业需要在所在地设区的市级药监局进行注册备案和生产许可备案,境外医疗器械生产企业需要委托境内代理人在国家药监局进行相应备案。
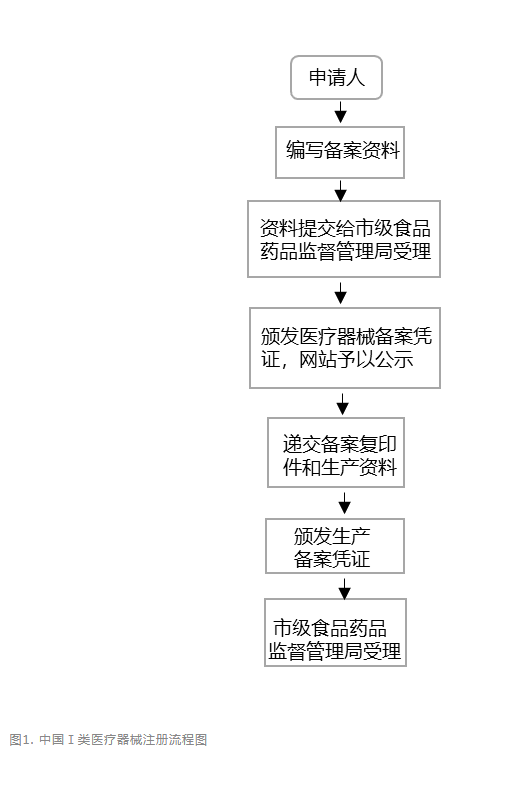
针对第Ⅱ类器械,境内生产企业需要在所在地省(直辖市)药监局进行注册申请、产品检验(经国家认可的检测机构监测)、质量体系考核申请、临床验证(是否实施需依据管理要求进行),境外医疗器械生产企业需要委托境内代理人在国家药监局进行相应注册。
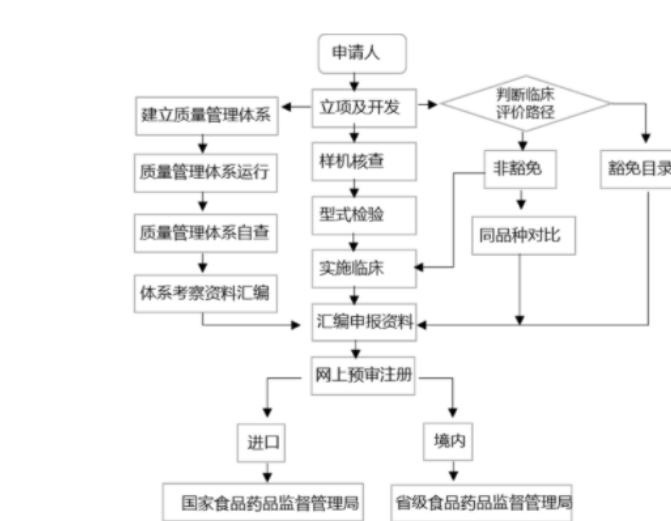
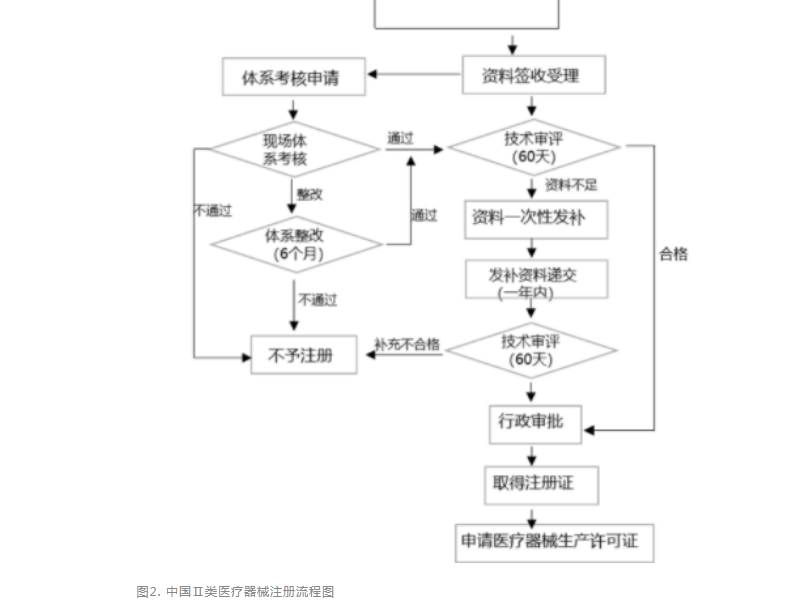
针对第Ⅲ类器械,无论境内、境外医疗器械生产企业均需要在国家药监局进行注册申请,产品检验(经国家认可的检测机构监测)、产品临床验证(如有规定可申请免临床)、质量体系考核申请。
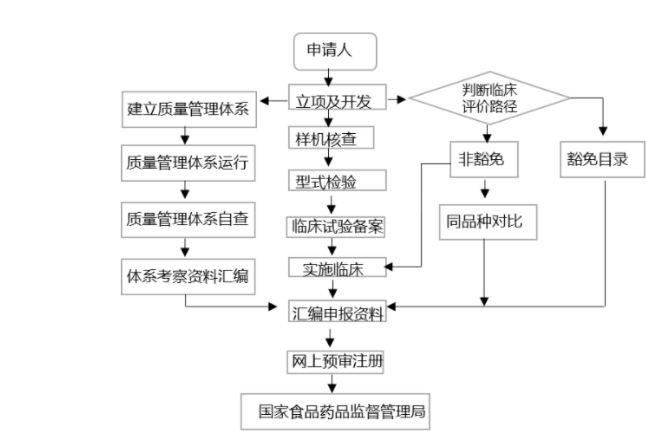
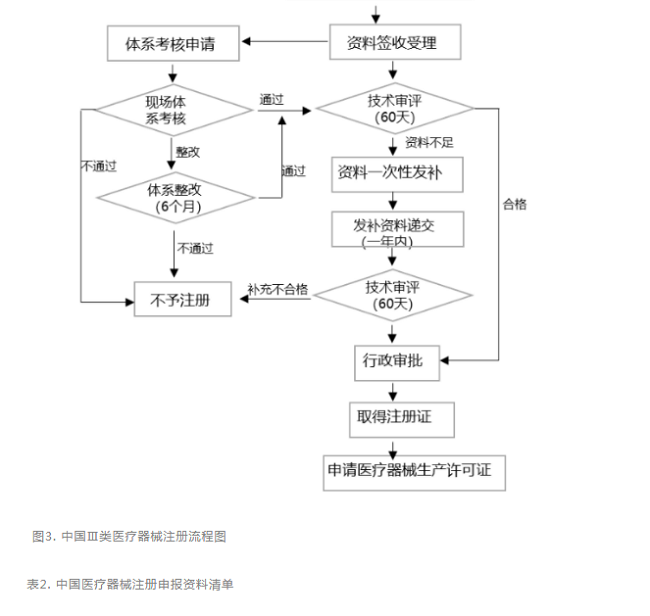
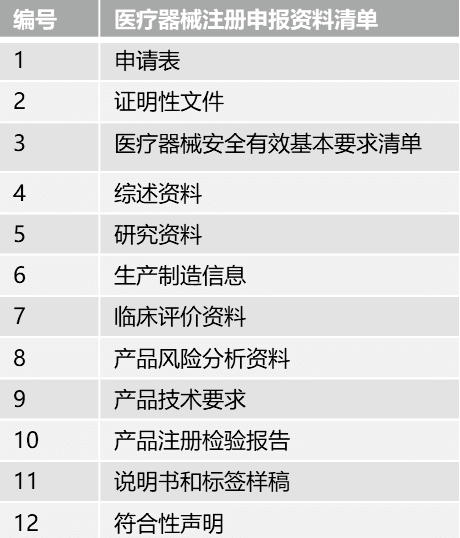
我国医疗器械行业起步晚,投入规模和核心技术与国际巨头存在一定的差距。行业呈现出“大而不强”的现状。为了摆脱这种状况,我国在近年愈发重视医疗器械行业创新。尤其在高端医疗器械领域,提出了“国产替代”的理念,并发布了创新医疗器械审批程序。这一措施对鼓励医疗器械的研究与创新,促进医疗器械新技术的推广和应用,推动医疗器械产业发展起到了积极作用。对于符合1、拥有产品核心技术发明专利权。2、产品主要工作原理或作用机理为国内首创,技术领先,安全性有根本性改进,具有显著的临床应用价值。3、具有基本定型的医疗器械,可以考虑申请创新医疗器械特别审批。国家食品药品监督管理总局医疗器械技术审评中心专门设立了创新医疗器械审查办公室并给与了一定的审批流程政策利好,如优先办理质量管理体系考核申请,制定专人进行沟通,提供指导,优先进行技术评审和行政审批。大致申请审批流程见图4。
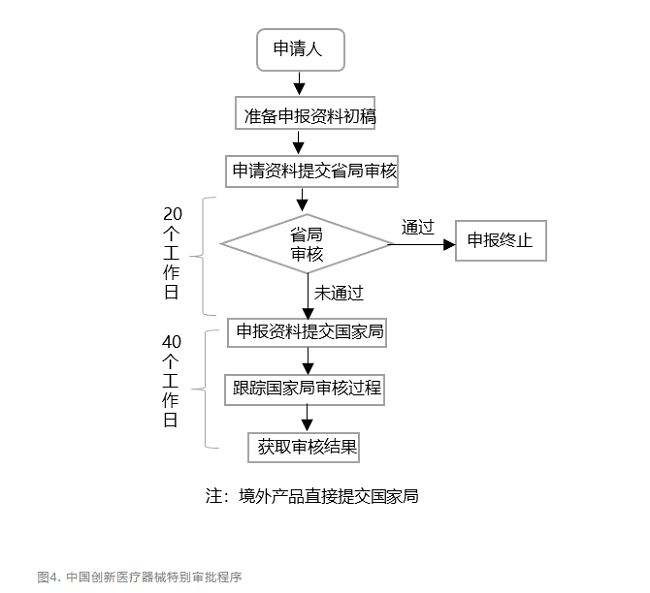
此外,需注意中国的注册证书具有5年的有效期。效期届满需要延续注册的,注册人应当在医疗器械注册证有效期届满6个月前,向药品监督管理部门申请延续注册,获得新的注册证书后产品才能继续上市销售。已注册的第二类、第三类医疗器械产品,其设计、原材料、生产工艺、使用范围、使用方法等发生实质性变化,有可能影响该医疗器械安全、有效的,注册人应向原注册部门申请办理变更注册手续。发生非实质性变化,不影响该医疗器械安全、有效的,应当将变化情况向原注册部门备案。
一、引言 自2019年新冠肺炎疫情发生以来,口罩在疫情防控中起着重要作用,科学佩戴口罩能有效预防新冠肺炎、流感等呼吸道传染病。同时,......
在我司专业人员的辅导下佛山客户取得29个型号规格的手动轮椅车注册证......
医疗器械经营许可证是医疗器械经营企业必须具备的证件,开办第二类医疗器械经营企业,应当向省、自治区、直辖市人民政府药品监督管理部门备案。......
2022年2月,广东省药品监督管理局共批准注册第二类医疗器械产品75个,其中首次注册19个,延续注册56个(具体产品见附件)。 特......
广州安思泰企业管理咨询有限公司是一家专业从事国内医疗器械行业注册咨询公司,为客户提供专业的咨询服务,公司奉行“围绕法规、标准,专业服务客户,......
按照《总局办公厅关于体外诊断试剂说明书文字性变更有关问题的通知》(食药监办械管[2016]117号),体外诊断试剂说明书信息性内容的文字......
......
广州安思泰企业管理咨询有限公司是一家专业从事国内医疗器械行业注册咨询公司,为客户提供专业的咨询服务,公司奉行“围绕法规、标准,专业服务客户,......
2019年5月我司顺利与天河区客户签订了二、三类经营许可证(包含体外诊断试剂,植入介入类)的项目......
为进一步深化医疗器械审评审批制度改革,依据医疗器械产业发展和监管工作实际,按照《医疗器械监督管理条例》《医疗器械分类目录动态调整工作程序》有......